Estimating the Population Frequency of a DNA Pattern
Next: Determining Allele Frequencies
Up: No Title
Previous: No Title
DNA "exclusions" are easy to interpret: if technical artifacts can be
excluded, a nonmatch is definitive proof that two samples had different
origins. But DNA "inclusions" cannot be interpreted without knowledge of
how often a match might be expected to occur in the general population.
Because of that fundamental asymmetry, although each new DNA typing
method or marker can be used for investigation and exclusion as soon as its
technical basis is secure, it cannot be interpreted with regard to inclusion
until the population frequencies of the patterns have been established. We
discuss the issues involved in estimating the frequency of a DNA pattern,
consisting of pairs of alleles at each of several loci.
Estimating Frequencies of DNA Patterns by Counting
A standard way to estimate frequency is to count occurrences in a random
sample of the appropriate population and then use classical statistical
formulas to place upper and lower confidence limits on the estimate.
Because estimates used in forensic science should avoid placing undue
weight on incriminating evidence, an upper confidence limit of the
frequency should be used in court. This is especially appropriate for
forensic DNA typing, because any loss of power can be offset by studying
additional loci.
To estimate the frequency of a particular DNA pattern, one might count the
number of occurrences of the pattern in an appropriate random population
sample. If the pattern occurred in I of 100 samples, the estimated frequency
would be 1%, with an upper confidence limit of 4.7%. If the pattern
occurred in 0 of 100 samples, the estimated frequency would be 0%, with
an upper confidence limit of 3%. (The upper bound cited is the traditional
95% confidence limit, whose use implies that the true value has only a 5%
chance of exceeding the upper bound.) Such estimates produced by
straightforward counting have the virtue that they do not depend on
theoretical assumptions, but simply on the sample's having been randomly
drawn from the appropriate population. However, such estimates do not
take advantage of the full potential of the genetic approach.
Estimating Frequencies of DNA Patterns with the
Multiplication Rule (Product Rule)
In contrast, population frequencies often quoted for DNA typing analyses
are based not on actual counting, but on theoretical models based on the
principles of population genetics. Each matching allele is assumed to
provide statistically independent evidence, and the frequencies of the
individual alleles are multiplied together to calculate a frequency of the
complete DNA pattern. Although a databank might contain only 500
people, multiplying the frequencies of enough separate events might result
in an estimated frequency of their all occurring in a given person of I in a
billion. Of course, the scientific validity of the multiplication rule depends
on whether the events (i.e., the matches at each allele) are actually
statistically independent.
From a statistical standpoint, the situation is analogous to estimating the
proportion of blond, blue-eyed, fair-skinned people in Europe by separately
counting the frequencies of people with blond hair, people with blue eyes,
and people with fair skin and calculating their proportions. If a population
survey of Europe showed that 1 of 10 people had blond hair, 1 of 10 had
blue eyes, and I of 10 had fair skin, one would be wrong to multiply these
frequencies to conclude that the frequency of people with all three traits was
1 in 1,000. Those traits tend to co-occur in Nordics, so the actual frequency
of the combined description is probably higher than 1 in 1,000. In other
words, the multiplication rule can produce an underestimate in this case,
because the traits are correlated owing to population substructure-the traits
have different frequencies in different population groups. Correlations
between those traits might also be due to selection or conceivably to the
action of some genes on all three traits. In any case, the example illustrates
that correlations within subgroups-whatever their origin-bear on the
procedures for estimating frequencies.
Unlike many of the technical aspects of DNA typing that are validated by
daily use in hundreds of laboratories, the extraordinary population-frequency
estimates sometimes reported for DNA typing do not arise in
research or medical applications that would provide useful validation of the
frequency of any particular person's DNA profile. Because it is impossible
or impractical to draw a large enough population to test calculated
frequencies for any particular DNA profile much below 1 in 1,000, there is not a
sufficient body of empirical data on which to base a claim that such
frequency calculations are reliable or valid per se. The assumption of
independence must be strictly scrutinized and estimation procedures
appropriately adjusted if possible. (The rarity of all the genotypes represented
in the databank can be demonstrated by pairwise comparisons. Thus, in a recently
reported analysis of the FBI database, no exactly matching pairs of profiles
were found in five-locus DNA profiles, and the closest match was a single
three-locus match among 7.6 million basepair comparisons.)[13]
The multiplication rule has been routinely applied to blood-group
frequencies in the forensic setting. However, that situation is substantially
different: Because conventional genetic markers are only modestly polymorphic
(with the exception of human leukocyte antigen, HLA, which
usually cannot be typed in forensic specimens), the multilocus genotype
frequencies are often about 1 in 100. Such estimates have been tested by
simple empirical counting. Pairwise comparisons of allele frequencies have
not revealed any correlation across loci. Hence, the multiplication rule does
not appear to lead to the risk of extrapolating beyond the available data for
conventional markers. In contrast, highly polymorphic DNA markers
exceed the informative power of protein markers, so multiplication leads to
estimates that are less than the reciprocal of the size of the databases.
Validity of Multiplication Rule and Population Substructure
The multiplication rule is based on the assumption that the population does
not contain subpopulations with distinct allele frequencies-that each
individual's alleles constitute statistically independent random selections
from a common gene pool. Under this assumption, the procedure for
calculating the population frequency of a genotype is straightforward:
- Count the frequency of alleles. For each allele in the genotype, examine a
random sample of the population and count the proportion of matching
alleles-that is, alleles that would be declared to match according to the rule
that is used for declaring matches in a forensic context. This step requires
only the selection of a sample that is truly random with reference to the
genetic type; it does not appeal to any theoretical models.
It is essential that the forensic matching rule be precise and objective-
otherwise it would be impossible to apply it in calculating the proportion of
individuals with matching alleles in the population databank. And it is
essential that the same rule be applied to count frequencies in the population
databank, because this is the only way to determine the proportion of
random individuals that would have been declared to match in the forensic
context. (In the context of forensic applications, an estimate of the
probability of a match in DNA typing has been termed conservative if on
the average it is larger than the actual one, so that any weight applied to the
estimate would favor the suspect. Thus, some laboratories use a more
conservative rule for counting population frequencies than for forensic
matches-an acceptable approach, because it overestimates allele
frequency. The converse would not be acceptable.)
- Calculate the frequency of the genotype at each locus. The frequency of a
homozygous genotype
 is calculated to be
is calculated to be 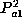 , where
, where 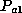 denotes the
frequency of allele
denotes the
frequency of allele 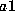 . The frequency of a heterozygous genotype
. The frequency of a heterozygous genotype 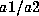 is
calculated to be
is
calculated to be  , where and
, where and 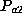 denote the frequencies
of alleles a1 and a2. In both cases, the genotype frequency is calculated by
simply multiplying the two allele frequencies, on the assumption that there
is no statistical correlation between the allele inherited from one's father and
the allele inherited from one's mother. The factor of 2 arises in the
heterozygous case, because one must consider the case in which allele al
was contributed by the father and allele
denote the frequencies
of alleles a1 and a2. In both cases, the genotype frequency is calculated by
simply multiplying the two allele frequencies, on the assumption that there
is no statistical correlation between the allele inherited from one's father and
the allele inherited from one's mother. The factor of 2 arises in the
heterozygous case, because one must consider the case in which allele al
was contributed by the father and allele 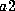 by the mother and vice versa:
each of the two cases has probability
by the mother and vice versa:
each of the two cases has probability 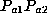 . When there is no correlation
between the two parental alleles, the locus is said to be in Hardy-Weinberg
equilibrium. We should note that in forensic DNA typing, a slight
modification is used in the case of apparently homozygous genotypes.
When one observes only a single allele in a sample, one cannot be certain
that the individual is a homozygote; it is always possible that a second allele
has been missed for technical reasons. To be conservative, most forensic
laboratories do not calculate the probability that the sample has two copies
of the allele (which is ), but rather the probability that the sample has at
least one copy (which is
. When there is no correlation
between the two parental alleles, the locus is said to be in Hardy-Weinberg
equilibrium. We should note that in forensic DNA typing, a slight
modification is used in the case of apparently homozygous genotypes.
When one observes only a single allele in a sample, one cannot be certain
that the individual is a homozygote; it is always possible that a second allele
has been missed for technical reasons. To be conservative, most forensic
laboratories do not calculate the probability that the sample has two copies
of the allele (which is ), but rather the probability that the sample has at
least one copy (which is 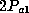 ) leaving open the possibility of a second
allele. We endorse this procedure.)
) leaving open the possibility of a second
allele. We endorse this procedure.)
- Calculate the frequency of the complete multilocus genotype. The
frequency of a complete genotype is calculated by multiplying the genotype
frequencies at all the loci. As in the previous step, this calculation assumes
that there is no correlation between genotypes at different loci; the absence
of such correlation is called linkage equilibrium. (Some authors prefer to
reserve the term linkage equilibrium for loci on the same chromosome and
to use the term gametic phase equilibrium for loci on different
chromosomes.) Suppose, for example, that a person has genotype
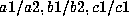 . If a random sample of the appropriate population shows that the
frequencies of
. If a random sample of the appropriate population shows that the
frequencies of  , and
, and  are approximately 0.1, 0.2, 0.3, 0.1,
and 0.2, respectively, then the population frequency of the genotype would
be estimated to be [2(0.1)(0.2)][2(0.3)(0.1)][(0.2)(0.2)] = 0.000096, or
about 1 in 10,417.
are approximately 0.1, 0.2, 0.3, 0.1,
and 0.2, respectively, then the population frequency of the genotype would
be estimated to be [2(0.1)(0.2)][2(0.3)(0.1)][(0.2)(0.2)] = 0.000096, or
about 1 in 10,417.
Again, the validity of the multiplication rule depends on the absence of
population substructure, because only in this special case are the different
alleles statistically uncorrelated with one another.
In a population that contains groups with characteristic allele frequencies,
knowledge of one allele in a person's genotype might carry some
information about the group to which the person belongs, and this in turn
alters the statistical expectation for the other alleles in the genotype. For
example, a person who has one allele that is common among Italians is
more likely to be of Italian descent and is thus more likely to carry
additional alleles that are common among Italians. The true genotype
frequency is thus higher than would be predicted by applying the
multiplication rule and using the average frequency in the entire population.
To illustrate the problem with a hypothetical example, suppose that a
particular allele at a VNTR locus has a 1% frequency in the general
population, but a 20% frequency in a specific subgroup. The frequency of
homozygotes for the allele would be calculated to be 1 in 10,000 according
to the allele frequency determined by sampling the general population, but
would actually be 1 in 25 for the subgroup. That is a hypothetical and
extreme example, but illustrates the potential effect of demography on gene
frequency estimation.
Basis of Concern About Population Substructure
The key question underlying the use of the multiplication rule is whether
actual populations have significant substructure for the loci used for
forensic typing. This has provoked considerable debate among population
geneticists: some have expressed serious concern about the possibility of
significant substructure,[2,4,9,10] and others consider the likely degree of
substructure not great enough to affect the calculations significantly.
[1,3,6,8,11-13]
The population geneticists who urge caution make three points:
1. Population genetic studies show some substructure within racial groups
for genetic variants, including protein polymorphisms, genetic diseases,
and DNA polymorphisms. Thus, North American Caucasians, blacks,
Hispanics, Asians, and Native Americans are not homogeneous groups.
Rather, each group is an admixture of subgroups with somewhat different
allele frequencies. Allele frequencies have not yet been homogenized,
because people tend to mate within these groups.
2. For any particular genetic marker, the degree of subpopulation
differentiation cannot be predicted, but must be determined empirically.
3. For the loci used for forensic typing, there have been too few empirical
investigations of subpopulation differentiation.
In short, those population geneticists believe that the absence of
substructure cannot be assumed, but must be proved empirically (see
Lewontin and Hartl [10]). Other population geneticists, while recognizing the
possibility or likelihood of population substructure, conclude that the
evidence to date suggests that the effect on estimates of genotype
frequencies are minimal (see Chakraborty and Kidd [12]). Recent empirical
studies concerning VNTR loci [13,14] (Weir, personal communication,
1991) detected no deviation from independence within or across loci.
Moreover, pairwise comparisons of all five-locus DNA profiles in the FBI
database showed no exact matches; the closest match was a single
three-locus match among 7.6 million pairwise comparisons.[13] These
studies are interpreted as indicating that multiplication of gene frequencies
across loci does not lead to major inaccuracies in the calculation of genotype
frequency-at least not for the specific polymorphic loci examined.
Although mindful of the controversy, the committee has chosen to assume
for the sake of discussion that population substructure may exist and
provide a method for estimating population frequencies in a manner that
adequately accounts for it. Our decision is based on several considerations:
1. It is possible to provide conservative estimates of population frequency,
without giving up the inherent power of DNA typing.
2. It is appropriate to prefer somewhat conservative numbers for forensic
DNA typing, especially because the statistical power lost in this way can
often be recovered through typing of additional loci, where required.
3. It is important to have a general approach that is applicable to any loci
used for forensic typing. Recent empirical studies pertain only to the
population genetics of the VNTR loci in current use. However, we expect
forensic DNA typing to undergo much change over the next decade-including
the introduction of different types of DNA polymorphisms, some
of which might have different properties from the standpoint of population
genetics.
4. It is desirable to provide a method for calculating population frequencies
that is independent of the ethnic group of the subject.
Assessing Population Substructure Requires Direct
Sampling of Ethnic Groups
How can one address the possibility of population substructure? In
principle, one might consider three approaches: (1) carry out population
studies on a large mixed population, such as a racial group, and use
statistical tests to detect the presence of substructure; (2) derive theoretical
principles that place bounds on the possible degree of population
substructure; and (3) directly sample different groups and compare the
observed allele frequencies. The third offers the soundest foundation for
assessing population substructure, both for existing loci and for many new
types of polymorphisms under development.
In principle, population substructure can be studied with statistical tests to
examine deviations from Hardy-Weinberg equilibrium and linkage
equilibrium. Such tests are not very useful in practice, however, because
their statistical power is extremely low: even large and significant
differences between subgroups will produce only slight deviations from
Hardy-Weinberg expectations. Thus, the absence of such deviations does
not provide powerful evidence of the absence of substructure (although the
presence of such deviations provides strong evidence of substructure).
The correct way to detect genetic differentiation among subgroups is to
sample the subgroups directly and to compare the frequencies. The
following example is extreme and has not been observed in any U.S.
population, but it illustrates the difference in power. Suppose that a
population consists of two groups with different allele frequencies at
a diabetic locus:

If there is random mating within the groups, Hardy-Weinberg equilibrium
within the groups will produce these genotype frequencies:
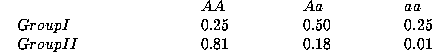
Suppose that Group I is 90% of the population and Group II is 10%. In the
overall population, the observed genotype frequencies will be
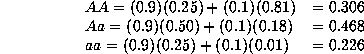
If we were unaware of the population substructure, what would we expect
under Hardy-Weinberg equilibrium? The average allele frequencies will be

which would correspond to the Hardy-Weinberg proportions of
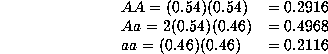
Even though there is substantial population substructure, the proportions do
not differ greatly from Hardy-Weinberg expectation. In fact, one can show
that detecting the population differentiation with the Hardy-Weinberg test
would require a sample of nearly 1,200, whereas detecting it by direct
examination of the subgroups would require a sample of only 22. In other
words, the Hardy-Weinberg test is very weak for testing substructure.
The lack of statistical power to detect population substructure makes it
difficult to detect genetic differentiation in a heterogeneous population.
Direct sampling of subgroups is required, rather than examining samples
from a large mixed population.
Similarly, population substructure cannot be predicted with certainty from
theoretical considerations. Studies of population substructure for protein
polymorphisms cannot be used to draw quantitative inferences concerning
population substructure for VNTRs, because loci are expected to show
different degrees of population differentiation that depend on such factors
as mutation rate and selective advantage. Differences between races cannot
be used to provide a meaningful upper bound on the variation within races.
Contrary to common belief based on difference in skin color and hair form,
studies have shown that the genetic diversity between subgroups within
races is greater than the genetic variation between races. [15] Broadly, the
results of the studies accord with the theory of genetic drift: the average
allele frequency of a large population group (e.g., a racial group) is
expected to drift more slowly than the allele frequencies of the smaller
subpopulations that it comprises (e.g., ethnic subgroups).
In summary, population differentiation must be assessed through direct
studies of allele frequencies in ethnic groups. Relatively few such studies
have been published so far, but some are under way.[16] Clearly, additional
such studies are desirable.
The Ceiling Principle: Accounting for Population Substructure
We describe here a practical and sound approach for accounting for
possible population substructure: the ceiling principled It is based on the
following observation: The multiplication rule will yield conservative
estimates, even for a substructured population, provided that the allele
frequencies used in the calculation exceed the allele frequencies in any of
the population subgroups. Accordingly, applying the ceiling principle
involves two steps: (1) For each allele at each locus, determine a ceiling
frequency that is an upper bound for the allele frequency that is independent
of the ethnic background of a subject; and (2) To calculate a genotype
frequency, apply the multiplication rule, using the ceiling frequencies for
the allele frequencies.
How should ceiling frequencies be determined? We must balance rigor and
practicality. On the one hand, it is not enough to sample broad populations
defined as "races" in the U.S. census (e.g., Hispanics), because of the
possibility of substructure. On the other hand, it is not feasible or
reasonable to sample every conceivable subpopulation in the world to
obtain a guaranteed upper bound. The committee strongly recommends the
following approach: Random samples of 100 persons should be drawn
from each of 15-20 populations, each representing a group relatively
homogeneous genetically; the largest frequency in any of these populations
or 5%, whichever is larger, should be taken as the ceiling frequency. The
reason for using 5% is discussed later.
We give a simplified example to illustrate the approach. Suppose that two
loci have been studied in three population samples, with the following
results:
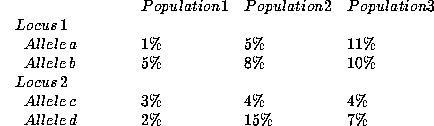
For the genotype consisting of a/b at locus 1 and c/d at locus 2, the ceiling
principle would assign ceiling values of 11% for allele a, 10% for allele b,
5% for allele c, and 15% for allele d and would apply the multiplication rule
to yield a genotype frequency of [2(0.11)(0.10)][2(0.05)(0.15)] =
0.00033, or about I in 3,000. Note that the frequency used for allele c is
5%, rather than 4%, to reflect the recommended lower bound of 5% on
allele frequencies. Because the calculation uses an upper bound for each
allele frequency, it is believed to be conservative given the available data,
even if there are correlations among alleles because of population
substructure and even for persons of mixed or unknown ancestry. This is
more conservative, and preferable, to taking the highest frequency
calculated for any of the three populations.
The ceiling principle reflects a number of important scientific and policy
considerations:
- The purpose of sampling various populations is to examine whether some
alleles have considerably higher frequencies in particular subgroups than in
the general population-presumably because of genetic drift. It is
matches at such alleles that might be accorded too much evidentiary weight,
if the general population frequency were used in calculating the probability
of a match.
- Determining whether an allele has especially high frequency does not
require a very large sample. A collection of 100 randomly chosen people
provides a sample of 200 alleles, which is quite adequate for estimating
allele frequencies.
- Genetically homogeneous populations from various regions of the world
should be examined to determine the extent of variation in allele frequency.
Ideally, the populations should span the range of ethnic groups that are
represented in the United States-e.g., English, Germans, Italians,
Russians, Navahos, Puerto Ricans, Chinese, Japanese, Vietnamese, and
West Africans. Some populations will be easy to sample through
arrangements with blood banks in the appropriate country; other
populations might be studied by sampling recent immigrants to the United
States. The choice and sampling of the 15-20 populations should be
supervised by the National Committee on Forensic DNA Typing (NCFDT)
described in Chapter 2.
We emphasize, however, that it is not necessary to be comprehensive. The
goal is not to ensure that the ethnic background of every particular
defendant is represented, but rather to define the likely range of allele
frequency variation.
- Because only a limited number of populations can be sampled, it is
necessary to make some allowance for unexamined populations. As usual,
the problem is rare alleles. Genetic drift has the greatest proportional effect
on rare alleles and may cause substantial variation in their frequency. Even
if one sees allele frequencies of 1% in several ethnic populations, it is not
safe to conclude that the frequency might not be five-fold higher in some
subgroups.
To overcome this problem, we recommend that ceiling frequencies be 5%
or higher. We selected this threshold because we concluded that allele
frequency estimates that were substantially lower would not provide
sufficiently reliable predictors for other, unsampled subgroups. Our
reasoning was based on population genetic theory and computational
results, and we aimed at accounting for the effects of sampling error and for
genetic drift. The latter consideration was especially important, because it
scales inversely with effective population size (i.e., small populations have
larger drift) and because it accumulates over generations. The use of such a
ceiling frequency would correspond to a lower bound of 5% on allele
frequencies. Even if one observed allele frequencies of about 1 %, one
would guard against the possibility that the frequency in a subpopulation
had drifted higher by using the lower bound of 5%. Thus, the lowest
frequency attributable to any single locus would be 1/400 (1/20 x 1/20). In
any case, it seems reasonable not to attach much greater weight to any
single locus.
- The ceiling principle yields the same frequency for a genotype, regardless
of the suspect's ethnic background, because the reported frequency
represents a maximum for any possible ethnic heritage. Accordingly, the
ethnic background of an individual suspect should be ignored in estimating
the likelihood of a random match. The calculation is fair to suspects,
because the estimated probabilities are likely to be conservative in their
incriminating power.
Some legal commentators have pointed out that frequencies should properly
be based on the population of possible perpetrators, rather than on the
population to which a particular suspect belongs.[17,18] Although that
argument is formally correct, practicalities often preclude use of that
approach. Furthermore, the ceiling principle eliminates the need for
investigating the perpetrator population, because it yields an upper bound to
the frequency that would be obtained by that approach.
Some have proposed a Bayesian approach,[19-21] to the presentation of
DNA evidence. However, this approach, focusing on likelihood ratios,
does not avoid the kinds of population genetic problems discussed in this
chapter. The committee has not tried to assess the relative merits of
Bayesian and frequentist approaches, because, outside the field of paternity
testing, no forensic laboratory in this country has, to our knowledge, used
Bayesian methods to interpret the implications of DNA matches in criminal
cases.
- Although the ceiling principle is a conservative approach, we feel that it is
appropriate, because DNA typing is unique in that the forensic analyst has
an essentially unlimited ability to adduce additional evidence. Whatever
power is sacrificed by requiring conservative estimates can be regained by
examining additional loci. (Although there could be cases in which the
DNA sample is insufficient for typing additional loci with RFLPs, this
limitation is likely to disappear with the eventual use of PCR.) A
conservative approach imposes no fundamental limitation on the power of
the technique.
Next: Determining Allele Frequencies
Up: No Title
Previous: No Title
laurie.snell@chance.dartmouth.edu